Effects of Past and Present-Day Landscape Structure on Forest Soil Microorganisms
 Sophie Mennicken1,2
Sophie Mennicken1,2  Floriane Kondratow3 Florian Buralli3
Floriane Kondratow3 Florian Buralli3  Sophie Manzi1 Emilie Andrieu3
Sophie Manzi1 Emilie Andrieu3  Mélanie Roy1*
Mélanie Roy1*  Antoine Brin3
Antoine Brin3- 1Evolution et Diversité Biologique (EDB), Université Toulouse 3 Paul Sabatier, CNRS, IRD, Toulouse, France
- 2Biology of Ecosystems Department, Faculty of Science, University of South Bohemia, Ceské Budejovice, Czechia
- 3Purpan School of Engineering, University of Toulouse, Toulouse, France
Theory surrounding landscape ecology has been built on the species distribution of birds and plants, but increasing evidence now exists for below-ground organisms, whose dispersal may also be affected by above-ground landscape structures. Uncertainties remain for how communities of microorganisms respond to landscape structure over time, and whether some groups of microorganisms react more than others. Here, we investigated if fungal or bacterial diversity is driven by the amount of forest cover in the current or the past landscape. We tested the habitat amount hypothesis (HAH) on ancient forests of the Cevennes national park, that experienced increased fragmentation 150 years ago, and are today surrounded by recent forests. As ancient forests are often more diverse in plant species, we hypothesized that the higher quantity of ancient forests in the landscape, the richer local fungal and bacterial communities would be. More precisely, we expected that ectomycorrhizal fungi, and pathotrophic fungi, often indicators of mature forests, would be also more sensitive to forest history and therefore to the quantity of ancient forests than bacteria and saprotrophic fungi. We sampled 40 soil cores per 0.5 ha, pooled in 8 composite samples per plot in 27 landscapes and sequenced ITS1 and 16S markers by Illumina-Mi seq. To identify functional groups of fungi, we relied on their taxonomy and the use of public databases. Our results partly follow the HAH, as fungal richness was positively related with the quantity of ancient forests in the landscape and not by the focal patch size. Ectomycorrhizal and pathotrophic fungi were positively affected by the ancient forest cover, and so were saprotrophic ones, but not bacteria. Local factors also shaped the communities such as soil composition and elevation, confirming classical patterns in soil microbial ecology. Interestingly, past landscape structure was better at explaining fungal community richness than contemporary landscape, suggesting a time lag in the response of communities to landscape modification and a potential extinction debt. Our results reveal the importance of below-ground communities in studies of landscape and historical ecology, with their structure and functions likely to be intimately linked with soil and landscape history.
Introduction
Soil microorganisms have only recently been included in studies of landscape ecology (Almasia et al., 2016; Grilli et al., 2017), with the discipline focusing mainly on plant, bird or insect species distributions and dispersal. Microorganisms such as fungi and bacteria, are among the most abundant and diverse organisms in soil (Fierer et al., 2007a) and play diverse and key roles in forest soil processes (e.g., decomposition, nutrient cycles, biotic relations; Altieri, 1999; Birkhofer et al., 2012; Hobara et al., 2014; Bani et al., 2018). Soil microorganisms are also intimately linked with plant communities, and could be affected by the landscape defined for plants, such as fields, forest, or hedgerows. However, for soil microorganisms, one major challenge in landscape ecology is to delineate patches, and corridors, depending on species dispersal abilities. For fungi, dispersal can occur over long distances (Vincenot and Selosse, 2017), but is often local (Peay et al., 2012), and thus size of patches may be extremely variable. Moreover, recent studies of landscape ecology on fungi have shown highly contrasting patterns, depending on the functional community and the scale studied (Nordén et al., 2018). For example, wood-associated fungi are distinct at forest edges and are affected by forest fragmentation (Helander et al., 2007; Abrego and Salcedo, 2014). Similarly, in the lava-fragmented landscape of Hawaï, local root-associated fungal diversity increased with forest area, and fungal species composition was correlated with fragment connectivity (Vannette et al., 2016). On the contrary, fragmentation did not shape communities of arbuscular mycorrhizal fungi associated to a ruderal forb (Grilli et al., 2015) while it affected ectomycorrhizal communities associated with Alnus, Crataegus and Corylus (Boeraeve et al., 2018a). Overall, Grilli et al. (2017) concluded that in general, fragmentation negatively affects fungal diversity but did not discuss other landscape characteristics. Among other soil microorganisms, bacterial distribution has already been revealed to respond to landscape effects (Fierer et al., 2007b; Culman et al., 2010; de Vries et al., 2012), at least in the context of streams, alpine soils and agricultural landscapes. Under more continuous cover such as forests, the response of bacteria to landscape has not been investigated and could reveal strong spatial patterns, as suggested by their high spatial turnover in heterogeneous environments (Horner-Devine et al., 2004).
Beyond fragmentation, the landscape can shape species distributions in various ways, with for example Fahrig (2013) suggesting that the quantity of habitat in the landscape could be more important than its spatial distribution. This hypothesis, named “habitat amount hypothesis” (HAH) has been intensely debated (Fahrig, 2015; Hanski, 2015) as for some authors it ignores well-known effects of landscape configuration (Haddad et al., 2017), and may not apply to the more isolated patches (Lindgren and Cousins, 2017) nor to all organisms. For example, it was verified for small mammals (Melo et al., 2017) but not for saproxylic beetles (Seibold et al., 2017). A meta-analysis based on 13 tests of the HAH suggested that patch isolation and size, after controlling for habitat amount, have an overall weak effect (Martin, 2018). The synthesis of Martin (2018) concluded that response to landscape configuration and HAH would be taxa- and context-specific, calling for repeated studies on a broader range of organisms. We thus decided to investigate the response of fungal and bacterial communities to the amount of available habitat, which could reveal alternative hypothesis when investigating the importance of fragmentation or patch area on these organisms. More precisely, we aimed to test if ancient forest quantity could shape existing fungal and bacterial communities.
The impact of historical patterns of habitat cover on contemporary biodiversity patterns has been documented in many fragmented landscapes (Harding et al., 1998; Kuussaari et al., 2009). Indeed, as it may take many generations for populations to go extinct after habitat loss and fragmentation, it has been hypothezised that a “ghost of land-use past” (Harding et al., 1998) can be a powerful driver of present-day diversity. If so, this would imply considering landscape history in conservation planning (Schrott et al., 2005; Kuussaari et al., 2009). A few pioneer studies have already demonstrated that fungal distribution is partly explained by ancient roman occupancy (Diedhiou et al., 2009), and that ectomycorrhizal fungi functions are distinct on such sites (Diedhiou et al., 2010). Case studies in Norway also revealed that the distribution of lichens – noted slow growing organisms- was shaped by the distribution of old oaks (Ranius et al., 2008a, b). Finally, Berglund and Jonsson (2005) revealed an extinction debt (Kuussaari et al., 2009) in lichens but not for wood-decaying fungi, suggesting that fungi with distinct ecologies may react differently to landscape dynamics and forest histories. Such evidence supports the consideration of both HAH and historical delineation of landscapes when investigating current soil microorganism distribution.
In temperate forests, ancientness, defined as the “temporal continuity of wooded soil” (Cateau et al., 2015), is a major characteristic of forests. Ancient forests are habitats for numerous species of plants (Hermy et al., 1999; Bergès et al., 2016; Bergès and Dupouey, 2017), and 57 to 132 species are recognized as indicators of ancient forests in France and Europe, respectively. While fungi can be indicator of forest maturity and therefore old-growth forests (Parmasto, 2001; Christensen et al., 2005; Dvořák et al., 2017; Halme et al., 2017), their link with ancient landscapes has received much less attention (Hofmeister et al., 2014). Interestingly, ancient forests of Belgium have revealed a higher diversity of fungi as compared with fragmented recent forests (Boeraeve et al., 2018b), but similar to non-fragmented forests, which suggests that both landscape structure and forest history may shape fungal diversity. To our knowledge, no study has investigated the bacterial communities of ancient forests. A recent study showed that bacterial communities are influenced by local abiotic factors, and not by soil history (Almasia et al., 2016). These results confirmed anterior studies that showed that pH was the best predictor for soil bacteria community distribution (Fierer and Jackson, 2006; Lauber et al., 2009). Investigating both fungi and bacteria in ancient forests may therefore reveal contrasting patterns, and we could expect fungi to be more specifically associated with ancient forests.
The development of both environmental sequencing to investigate soil organisms (Taberlet et al., 2012; Zinger et al., 2016) and public databases of historical maps (Bergès and Dupouey, 2017) make such a study of soil community and landscape ecology possible. Indeed, environmental sequencing can target all bacteria and fungi with universal markers, and enable the sequencing of both ectomycorrhizal, saprotrophic or pathotrophic ones. In parallel, public databases are now reporting functional traits for hundreds of soil taxa (Nguyen et al., 2016), and can therefore be used to derive functional diversity from environmental sequencing. Until now, wood associated species, often pathotrophs, are recognized as indicators of old-growth forests (Parmasto, 2001; Dvořák et al., 2017; Halme et al., 2017) and the question remains as to whether such species are also sensitive to the quantity of ancient forests in the landscape. Moreover, as ectomycorrhizal fungi are intimately associated with trees, we could hypothesize that they would also be favored by forest continuity and the quantity of ancient forests in the landscape. Conversely, for saprotrophic fungi, abundant in the litter, and bacteria, the question remains as to whether they would reflect differences in landscape structure in a forest context.
In Cevennes National Park, ancient forests are scattered in the landscape, and occur surrounded by conifer plantations, or recently established beech (Fagus sylvatica) forests. While 80% of the Park territory is represented by forests today, only 13% of the territory was covered by forests in 1850 (Figure 1). The difference between ancient and recent forest distribution is particularly high as compared to other forests in France (Bergès and Dupouey, 2017), and represents a study site to investigate the response of soil microorganisms to past and present landscapes, and test HAH. To better understand the effect of landscape on below-ground communities, we therefore handled a large-scale sampling of soil communities in Cevennes National Park. Our objectives were to test (1) if fungi and bacteria communities could follow the HAH, being richer in forests surrounded by higher forest cover, (2) if functional groups show distinct responses to the landscape structure, and (3) if soil microorganism communities show a response time lag to modification of landscape structure.
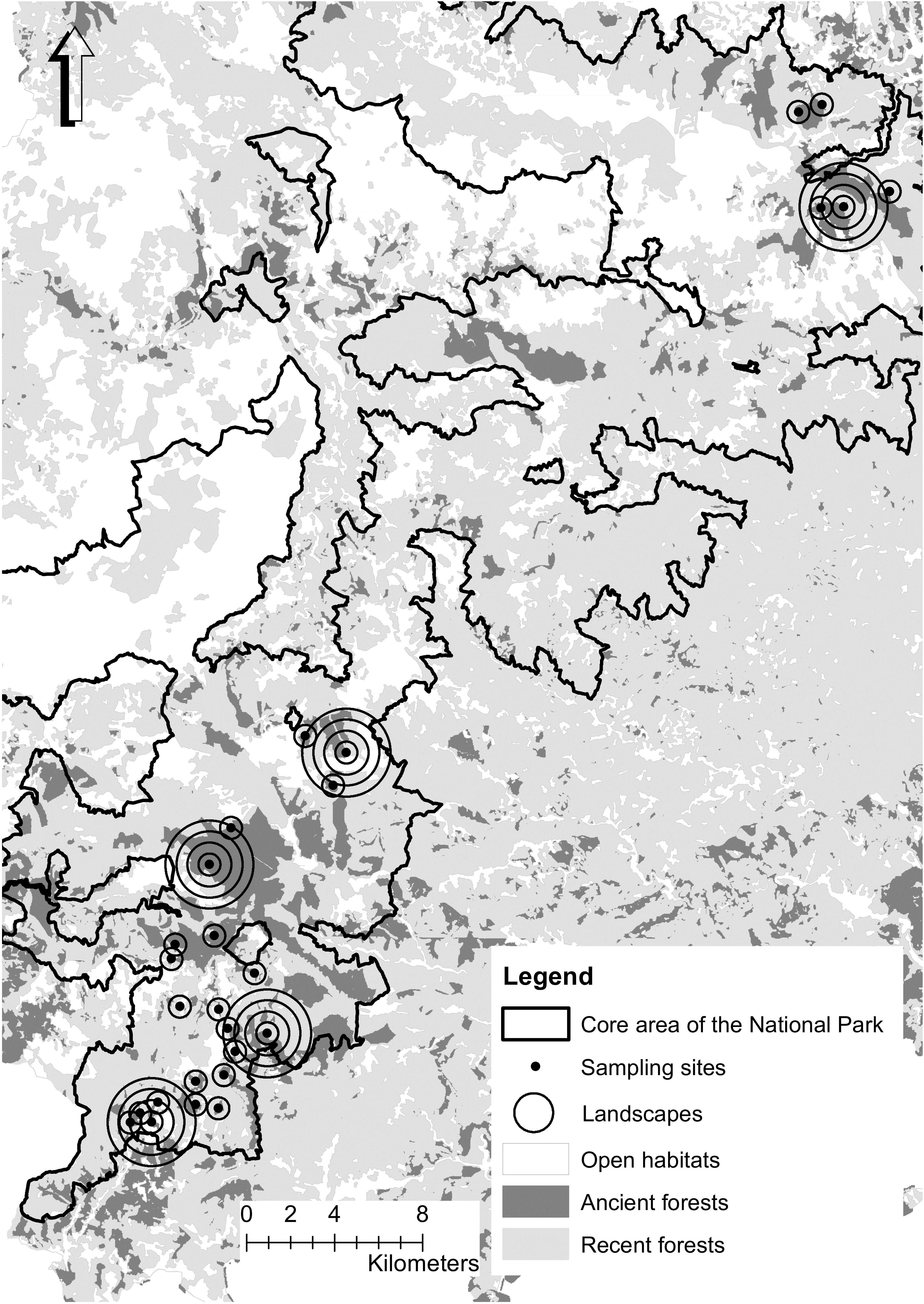
Figure 1. Map of Cevennes National Park and distribution of ancient forests and sampling sites. Landscapes are illustrated as circles of different radii around sampling plots, but for more clarity, only radii of 500 m are drawn for most sites.
Materials and Methods
Study Sites and Landscape Variables Design
The study area covers circa 15,000 hectares in the core area of Cevennes National Park (93,800 hectares). This area was mainly covered by forest (70% surface cover), dominated by European beech (F. sylvatica L.), mixed either with deciduous species (Quercus sp.) or with coniferous species (Abies alba Mill. and Picea abies (L.) Karst). We selected 27 landscapes to include a gradient of ancient forest cover (Figure 1). Following Fahrig (2013), we checked that the patch surface (including the sampling site) and the total surface occupied by ancient forests in the landscape were decorrelated (e.g., of patches, Figure 2, Supplementary Figure S1). Sample sites were located at the center of each landscape, in ancient forests dominated by beech, covering an area of 0.5 ha. The information on beech coverage and forest surface (both ancient and recent) were acquired from conservation maps (2011) provided by the National Park and based on digitized maps produced between 1920 and 1966 (IGN, 2011). Beech coverage was confirmed by an estimation of beech total basal area with a relascope at each site. We computed this information for buffers in a radius of 500, 1000, 1500, and 2000 meters around the central sampling patch (e.g., Figure 1). All landscapes were selected from the montane bioclimatic zone, between 935 and 1453 m a.s.l. and on granitic bedrock using MNT BD Alti® (IGN, 1996) and a geological map (Carte lithologique simplifiée®, BRGM, 2008). Total ancient forest area was calculated for landscapes of 78, 314, 706, and 1256 hectares.
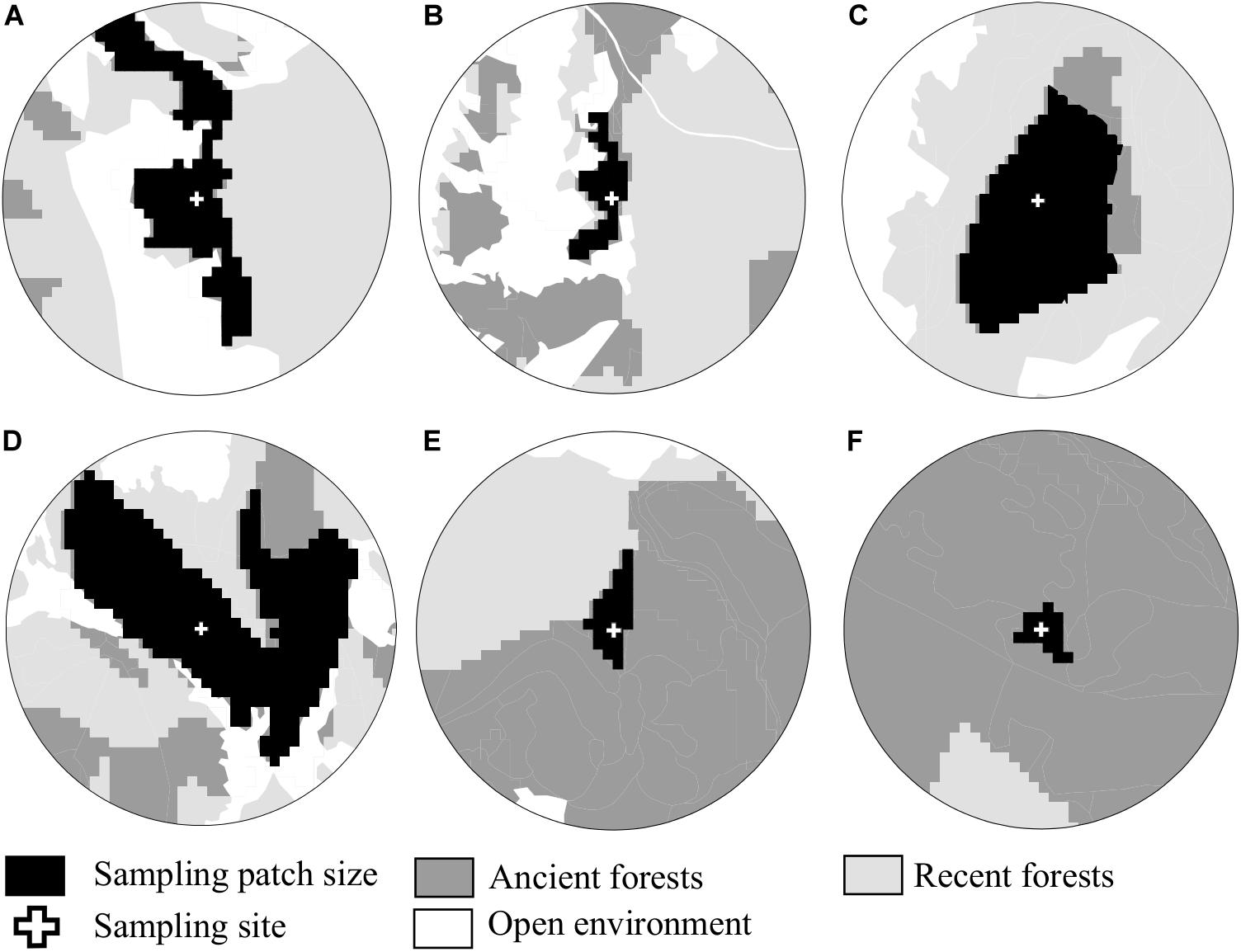
Figure 2. Examples of landscape structure, illustrating gradients of habitat amount in the landscape, from (A) a landscape dominated by recent forest, to (F) a landscape dominated by ancient forest. The size of the central patch can be variable, and does not correlate with the quantity of ancient forest in the buffer. As an example, both large (A–C) and small patches (B,E,F) of ancient forests were sampled, in landscapes with a variable quantity of ancient forests (increasing from (A–F)).
Soil Sampling and Processing
Soil samples were collected during April 2017 (average temperature 4.4°C, average precipitation 17.6 mm from www.infoclimat.fr). At each study site, soil cores were collected along two 60 m transects, spaced 20 m apart. Every 20 m along a transect, a soil core was collected, and pooled with four soil cores collected at 3.5 m from the focal point in each cardinal direction. The soil core itself was collected with a shovel, from the first 10 cm of soil depth, targeting the A horizon and removing the larger debris from the litter. Between each soil core, a false core was sampled in a radius a 1m, to remove the debris from the previous sampling point, and prepare the next sampling point. The shovel was clean every night with 2% bleach and rinsed with tap water (i.e., every two plots). For each plot, a total of 40 soil cores were then collected and pooled into 8 composite samples, before being returned to the lab the same day. 15 g of each composite sample was isolated in clean tea-bags and dried with 50 g of silicagel (with more added if the sample was not dry the day after). Soil DNA was extracted less than 1 month later from the dry samples (circa 10 g of soil) with Macherey Nagel NucleoSpin® Soil kit (Ref 740780.250). Amplifications targeted ITS1 nuclear rDNA primers for fungi (Fwd: ITS5 GGAAGTAAAAGTCGTAACAAGG from Epp et al., 2012 and a modified version of Rev: 5.8S_Fungi CAAGAGATCCGTTGTTGAAAGTK, Taberlet et al., 2018), and 16S rDNA (V5,V6) for bacteria [Bact01 primers – Fwd: GGATTAGATACCCTGGTAGT and Rev: CACGACACGAGCTGACG (Fliegerova et al., 2014)]. Each PCR was performed with a unique set of primers, characterized by a unique set of 8 nucleotide tags added to the universal primers, to distinguish sample sequences later in the analysis (as described in Taberlet et al., 2018). Negative controls without any PCR reactants were included (representing 10% of our reactions) and all PCR reactions were replicated twice, each time with distinct primer pairs. Replicates were not amplified on the same PCR plate. PCR conditions followed Nagati et al. (2018). All PCR reactions were pooled to prepare a unique Illumina-Miseq library, as the use of 8 nucleotide tags allowed to distinguish replicates and samples sequenced within the library. Lastly, the library was sent to the GenoToul Core Facility platform for sequencing in two directions and at 250 bp (2∗250 bp).
Bioinformatic and Sequence Analysis
Bioinformatic analyses were handled with OBItools (Boyer et al., 2016). First, R1 and R2 reads were “paired-end” based on a 50 bp overlap, then low quality sequences, sequences shorter than 100 bp, and non-assembled sequences were removed. Sequences were attributed to samples thanks to their unique primer and 8-nucleotide tag combinations. Non-attributed sequences were discarded after this step. To speed up analyses, sequences were dereplicated, i.e., for each repeated sequence, one sequence was kept, and its occurrence per sample was counted. At this step sequences only occurring once in the dataset were removed, since these likely represented sequencing artifacts. Pairwise distances were then computed using sumaclust function to delineate sequence clusters with similarity of 97% or above (representing operational taxonomic units, OTUs, following Nilsson et al., 2008). Taxonomic assignment of OTUs was conducted using reference databases with the ecotag function (Boyer et al., 2016), which uses global alignment of sequences against full-length references. This function compares sampled short sequences with a database of short sequences, and returns not only the most similar sequence but also the taxonomic assignment of other closely similar sequences. With this method, low similarity sequences are assigned to “Fungi”, while sequences with a high similarity to several Russulaceae are assigned to “Russulaceae,” or even to an individual species. For fungi the reference database of short sequences was obtained by running an in silico PCR with the ecoPCR program (Ficetola et al., 2010) on Genbank (release 197;1) using the primer pairs employed here. Chimeras have very bad results with ecotag, as sequences are not assigned to a high taxonomic level. Then, taxonomic assignment yielding the highest similarity score was kept. For Bacteria, taxonomic assignment was not handled with ecotag but conducted using the SILVA database (Quast et al., 2012), with similarity parameters kept at 0.97. Subsequent dataset processing and analyses were conducted using R (R Core Team, 2019). Based on taxonomy, we removed all non-fungi and non-bacteria sequences, respectively. For bacteria, we also removed chloroplast or mitochondrial sequences. Using the sequenced negative controls, we also removed sequences abundant in the controls and rare in our samples (representing less than 5% of their counts). We had no case of sequences which were both abundant in samples and in controls. Finally, fungal guilds were assigned using FUNGUILD (Nguyen et al., 2016). Raw sequences were deposited on DRYAD2.
Soil Sampling for Chemical Analyses
After collecting soil for the DNA extraction, the remaining 485 g of soil of each composite sample was transferred to a paper bag and dried at 50°C for three days. Before drying, pH units were obtained by pH colorimetric measurement. All composite samples were used for soil chemical analysis, resulting in 8 samples per plot. The soil physicochemical analyses were performed according to current standards by a soil testing laboratory3 : total soil organic carbon content (NF ISO 10694), total nitrogen (NF ISO 13878), phosphorus (Duchaufour), measures of cation exchange capacity and exchangeable cations: calcium, magnesium, sodium, potassium, iron, aluminum and manganese (cobalt hexamine).
Data Analyses
All analyses were carried out with the R 3.6.1 (R Core Team, 2019) software, using the iNext package (Hsieh et al., 2016) to compute Hill numbers and sample coverage, the lme4 (Bates et al., 2015) and MuMin (Bartoñ, 2019) packages for the GLMMs, and the vegan package (Oksanen et al., 2019) for multivariate analysis.
Bacterial and Fungal Diversity and Sampling Coverage
First of all, to investigate if the NGS approach had produced comprehensive inventories, we assessed the sampling coverage at the site level using the framework proposed by Chao and Jost (2012). The sampling coverage was measured for each functional and taxonomic group.
Habitat Amount Effect on Species Diversity
To test the HAH, we investigated several indices of diversity via the Hill numbers framework (Hill, 1973; Alberdi and Gilbert, 2019) which allows the weighting of rare species through the parameter q. The larger the q value, the higher the importance attributed to abundant OTUs. We considered three values: q = 0 (equivalent to species richness), q = 1 (equivalent to Shannon index) and q = 2 (equivalent to the inverse of Simpson index). Diversity was computed separately for all bacteria and all fungi. For the latter, we also estimated diversity indices for each trophic guild (symbiotrophs, pathotrophs and saprotrophs).
To test if species diversity was positively correlated with ancient forest coverage in the landscape, we used GLMM with a Poisson distribution and several co-variables: the focal patch area, the first three axis of a PCA on edaphic descriptors (Table 1) and elevation. The site was included in our models, as a random effect, to account for spatial structure of the sampling design.
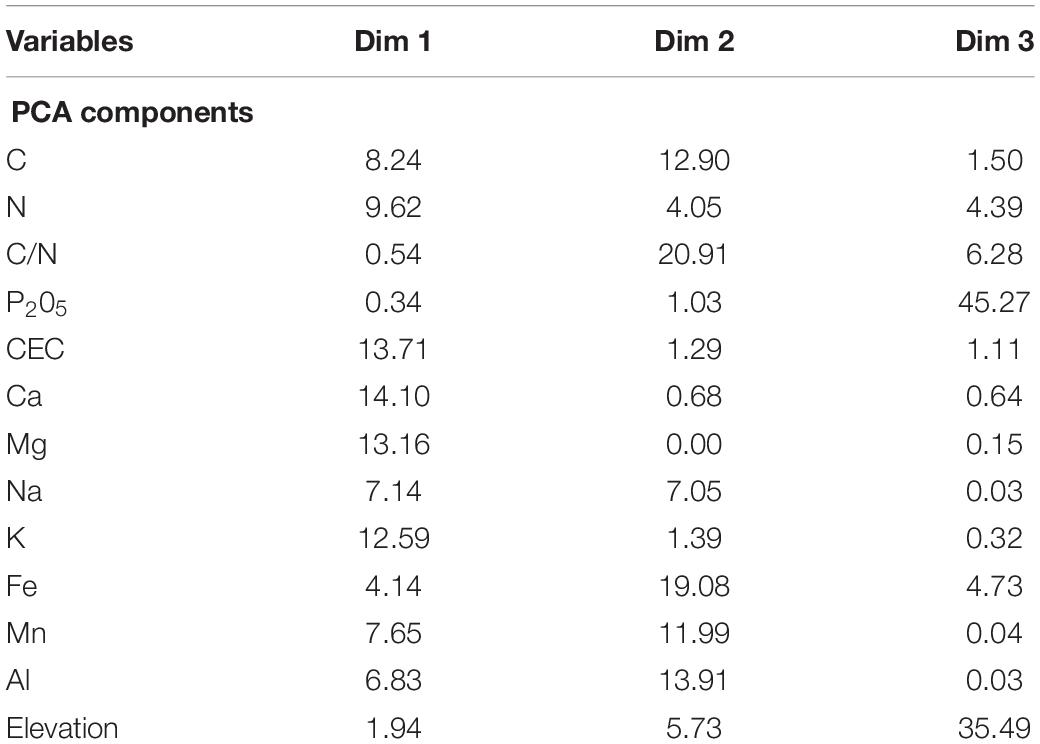
Table 1. Variable contributions (%) to the first three axis of the PCA of local descriptors.
Habitat Amount Effect on Community Composition
The effects of landscape structure and local descriptors on community composition were assessed by distance-based redundancy analysis. We used the Bray-Curtis dissimilarity index as recommended by Lucas et al. (2017) to analyze microbial communities described by high-throughput molecular techniques. To partial out spatial variation in community composition, we used the vectors created by a Principal Coordinates of Neighborhood Matrix (PCNM, Dray et al., 2006). Indeed, our sampling was scattered in Cevennes National Park, and we used this technique not to test the spatial structure, but to remove the variation linked to spatial structure and focus on habitat amount variations. Permutation tests were used to assess the significance of marginal effect of each predictor on Bray-Curtis distances.
Spatial and Temporal Scale of Effect
We conducted a multi-scale analysis to determine the optimal spatial extent (scale of effect) at which landscape structure (here the habitat coverage) best predicts our response variables (Brennan et al., 2002; Jackson and Fahrig, 2015). We considered four radii (500, 1000, 1500, and 2000 m, see Figure 1) for the landscape structure and estimated the scale of effect for each response variable by model comparison based on the small-sample Akaike Information Criteria (AICc). The scale of effect was the radius of the buffer (landscape) corresponding to the best model, chosen with the smallest AICc. Finally, to explore a legacy effect on current OTU diversity, we compared models built with either ancient or contemporary forest cover as habitat amount, at the relevant scale of effect for each response variable (Figure 3).
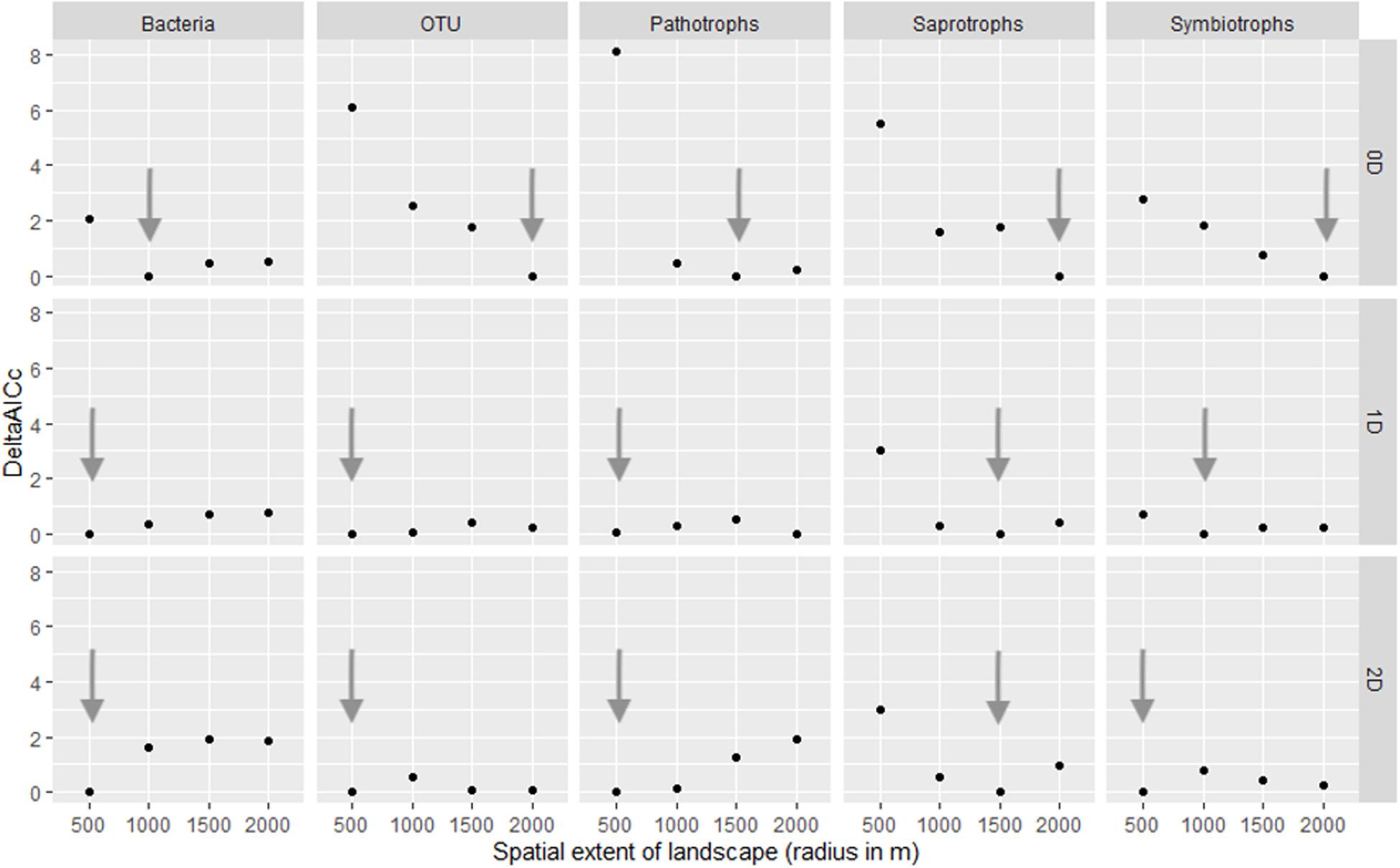
Figure 3. Estimation of the scale of effect of the landscape variable (habitat amount) for each group and each Hill number (0D: species richness, 1D: Shannon index and 2D: inverse of Simpson index). Arrows indicate the distance with the lower AIC and therefore the best model. OTU refer to all fungi OTUs.
Results
Bacterial and Fungal Diversity and Sampling Coverage
The sequencing run produced 3 014,848 and 2 137,868 sequences for fungi and bacteria, respectively (without singletons and low-quality sequences). After cleaning the sequences (using negative controls and taxonomic assignment), we detected 3 751 fungal OTUs from 1.31 × 106 sequences and 14 922 bacterial OTUs from 8.44 × 105 sequences. The number of sequences was lower for bacteria as sequences attributed to mitochondria and chloroplasts were removed. Rare OTUs, well identified to fungi and bacteria, represented a minor part of the dataset for fungi (10% of OTUs occurring once, 12% occurring twice), but a larger part of bacteria (22% occurring once, 22% occurring twice). For fungi, 36.5%, 27.4% and 9% of OTUs were unambiguously assigned to symbiotrophs, saprotrophs and pathotrophs, respectively. Other OTUs were either attributed to two trophic modes (e.g., pathotroph and saprotroph) or not attributed to a trophic mode given the low taxonomic resolution (for Dikarya sequences, i.e., 11.8% of OTUs). Ectomycorrhizal fungi represented 92.4% of symbiotroph fungi, while arbuscular mycorrhizal fungi represented 4.3% of OTUs. Other mycorrhizal fungi belonged to several guilds (e.g., ectomycorrhizal and orchid mycorrhizal or ericoid mycorrhizal), to endophytes (0.01%) or lichens (1.3%). Ectomycorrhizal OTUs were more diverse, with 1245 OTUs, mainly belonging to Russulaceae, Cortinariaceae and Atheliaceae (Supplementary Figure S2). Cortinariaceae were more abundant in sequences but Russulaceae were richer in OTUs (Supplementary Figure S2). Among bacteria, proteobacteria, acidobacteria and actinobacteria were dominant, with actinobacteria represented by more sequences and proteobacteria by more OTUs than other phyla (Supplementary Figure S3).
With a mean sampling coverage of 94% and 99% for bacteria and fungi, respectively, our sampling is comprehensive at the site level (Supplementary Figure S4). Communities were relatively rich at each site, with at least 445 fungal and 2030 bacterial OTUs at each site (maximum of 1121 and 3948 OTUs, respectively). For fungi, when looking at the trophic group level, the sampling coverage was 99%, 98% and 95% for symbiotrophs, saprotrophs and pathotrophs, respectively (Supplementary Figure S4). Furthermore, the sampling coverage was not affected by the number of reads, which suggests that the sampling coverage was high even locally for composite sample. Therefore, we did not have to include the number of reads as a covariable in our models.
Habitat Amount Effect on Species Diversity
PCA was used to reduce the number of soil variables included in the GLMMs. The first three axis of the PCA on soil variables summarized 82.5% of total inertia: the first axis was mainly explained by Ca, Mg and K contents together with the CEC, the second axis was mainly explained by C/N and Fe content, and the third axis was mainly explained by P2O5 content and elevation (Table 1).
The use of Hill number allowed to compare the response of all fungi including rare species, more common species and the more abundant ones. In line with our predictions, habitat coverage was significantly and positively related to fungal OTU richness and with OTU richness of the three fungal trophic groups (Table 2) while patch area had no effect on OTU diversity. Such pattern did not emerge among fungi for common OTUs (1D) and dominant OTUs (2D). However, the patch area was significantly and positively related to the Simpson diversity (2D) of saprotrophs (Table 2). Regarding bacteria, whatever the Hill number considered, we did not detect any significant relationship between diversity and habitat amount or patch area.
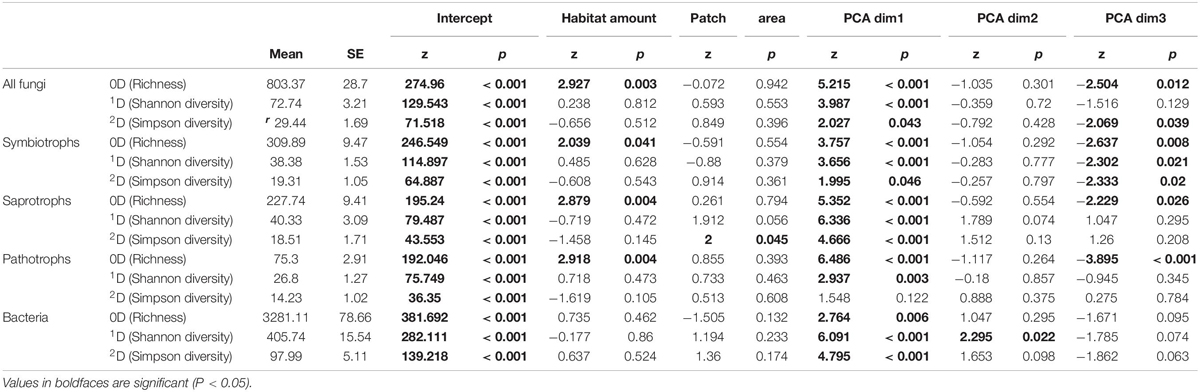
Table 2. Results from regression analyses.
On the other hand, for all taxa and trophic groups, the first component of PCA was the main driver of OTU diversity (Table 2). This axis was mainly driven by soil CEC, other cations concentration (Ca, Mg, K). The second axis, driven by Fe concentration and C/N ratio, only correlated with bacterial Shannon diversity. The third axis, mainly driven by phosphate concentration and elevation, was significantly and negatively related with OTU richness of all fungi, and each trophic group, but not for bacteria (Table 2). Whereas this correlation was also observed for common and dominant symbiotrophic OTUs, no relationship emerged for pathotrophs and saprotrophs when decreasing the importance of rare species on diversity indices (Table 2). Finally, for all indices and taxonomic or functional groups, the effect of PCA axis was always more important and significant than the habitat amount effect, showing that both factors contribute to shape microorganism diversity but that soil factors are also extremely important locally.
Habitat Amount Effect on Community Composition
The composition of communities was only affected by the three PCA axis but not by the habitat amount nor the patch area (Table 3). All fungal, symbiotrophic and saprotrophic communities were shaped by the three PCA axis, while bacterial communities were shaped by the two first axis, and pathotrophic fungi only by the first one.
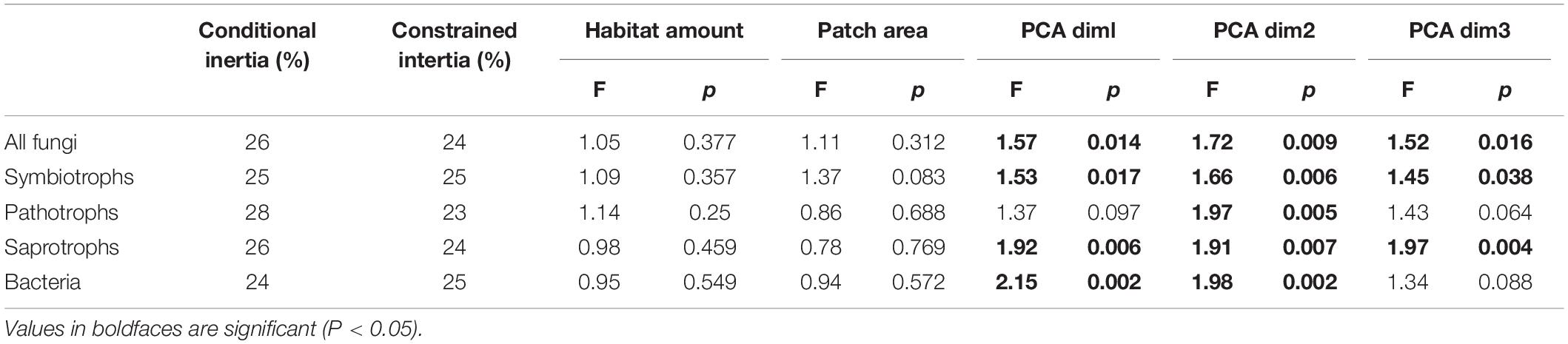
Table 3. Results from distance-based redundancy analysis (db-RDA) using Bray-Curtis dissimilarity.
Spatial Scale of Effect
The scale of effects (of the habitat amount) was variable according to Hill numbers and the group considered (Figure 3). As a general pattern, the scale of effect increased with increasing the weight of rare species in diversity indices (Figure 3, see arrows). Our landscapes were initially delineated in a radius of 500 m, which seems relevant to explain variations in 2D Hill number (inverse of Simson index) of all micro-organisms but saprotrophic fungi. Variation in species number, on which HAH was based, are on the contrary better explained by landscape structure delineated with a radius greater than 500 m (at least for all fungal OTUs, saprotrophic and symbiotrophic ones).
Ancient vs Actual Forest Cover
For all models with a significant effect of habitat amount, the models based on ancient forest cover had lower AICc than those based on contemporary forest cover (Table 4). This pattern was observed for all taxonomic and functional groups, but the difference of AICc was more important for saprotrophs and pathotrophs.
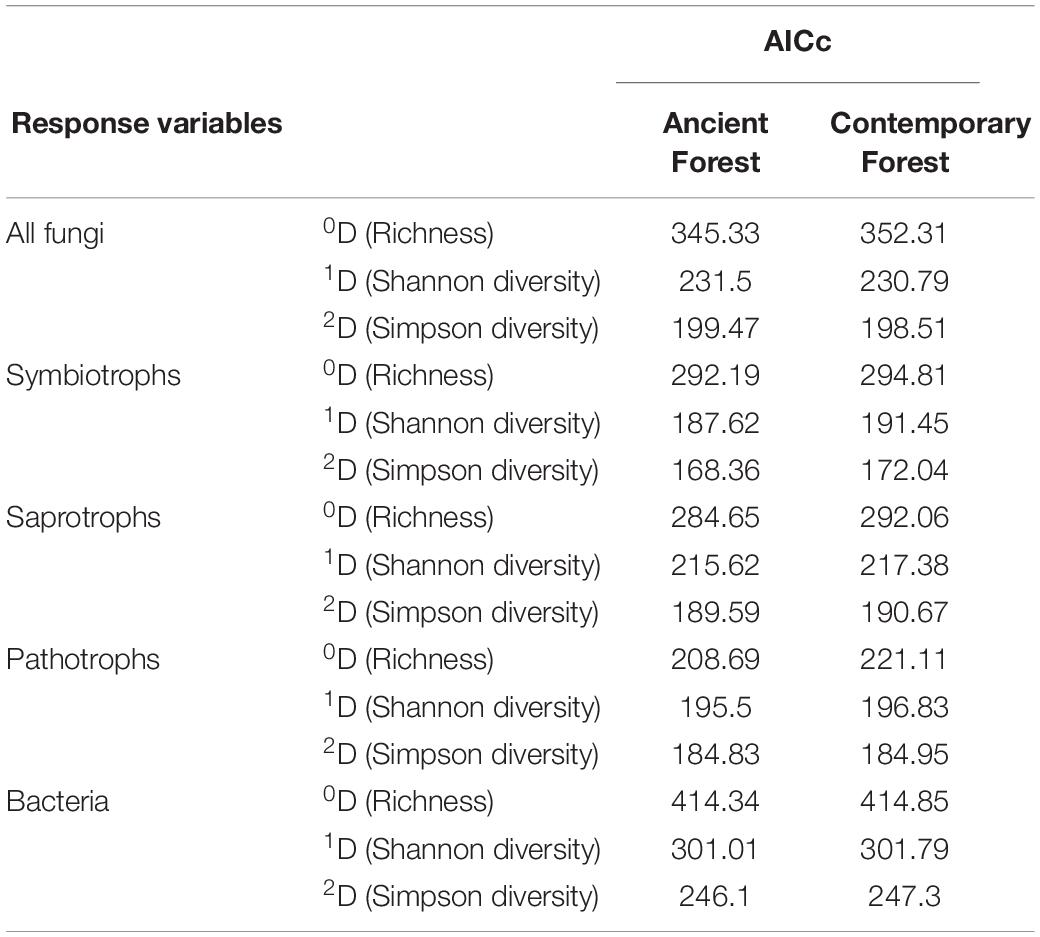
Table 4. Models comparisons, with AICc, when using either ancient or contemporary forest cover as habitat amount.
Discussion
While there are debates surrounding how fungi can be affected by fragmentation (Grilli et al., 2017), our study showed, for the first time, that habitat coverage in the landscape is a driver of the species richness of local communities in forest patches. We did not confirm this pattern for bacteria, nor for the more abundant species (according to Hill numbers), but showed that both ectomycorrhizal (the main symbiotrophic fungi in our analysis), pathotrophic and saprotrophic fungi responded to the amount of ancient forest in the landscape. This common trend for fungi probably contrasts with the differences in dispersal strategy among fungi (Douhan et al., 2011). However, all agree that most spores disperse locally (Adams et al., 2013). This result follows the observations of Peay et al. (2012), who measured the dispersal of ectomycorrhizal fungi from 1 m to 10 km, and showed a rapid decrease in colonization success with increasing distance to the source. Edman et al. (2004) also confirmed that local sources were particularly important for wood decaying fungi and recommended maintaining local sources of dead wood, paying attention to the vicinity of forest patches. Interestingly, this trend was not confirmed when decreasing the weight of rare species (see results for Shannon and Simpson indices), suggesting that rare species would be more sensitive to the habitat coverage and changes in landscape structure. For wood-inhabiting species, a similar pattern has been observed, and Nordén et al. (2013) showed that rare specialist fungi were more limited by their habitat loss than generalist fungi. As the importance of rare species may also be lowered with Bray-Curtis distances, this would also explain why we did not detect any relationship between habitat coverage and the composition of communities.
Conversely, the diversity of bacterial communities was not affected by the habitat coverage of ancient forests, but rather shaped by local environmental factors. While the habitat amount hypothesis has never been investigated per se on bacterial communities, early studies have already shown the importance of habitat fragmentation and patch size in experimental conditions (Burkey, 1997; Gilbert et al., 1998). Interestingly, a recent study of fragmented landscapes in Chile (Almasia et al., 2016) also highlighted the importance of local environmental factors but not historical factors, confirming that present day conditions would be more important than historical ones. This lack of response by bacteria to ancient forest quantity could also be explained by their high turnover and short life span. Some bacteria are able to be dormant (Kaprelyants et al., 1993), but this trait is not widespread, and might have been selected rather in extreme environments or at high elevations (Jones and Lennon, 2010; Pozzi et al., 2015). In the Cevennes national park, soil conditions are not extreme and may not select for dormant bacteria, which may reduce their sensitivity to past forest conditions. When considering bacterial dispersal, several studies agree that terrestrial bacteria are able to disperse over relatively long distances (Smith et al., 2013), greater than fungi (Chemidlin Prévost-Bouré et al., 2014; Vacher et al., 2016), but are still locally limited by environmental conditions (Horner-Devine et al., 2004). Our study corroborates the importance of local soil parameters in shaping bacterial diversity without definitively rejecting the HAH, which could be investigated in more contrasted soil landscapes (e.g., agricultural vs forest soils).
By revealing that past forest cover better explains variation of species diversity than recent forest cover does, our study raises questions on the persistence of past landscape structure and its effect. Numerous clues have accumulated for plant communities (Nordén et al., 2014), even in Amazonian forests where botanists have now revealed that plant diversity is higher close to pre-Colombian sites (Levis et al., 2017). Such legacy effects are therefore acknowledged for plants, especially for trees due to their long lifespan. For fungi, the persistence of effects raises questions, for example on fungal longevity in soil. Estimating the age of fungal genets is rarely done, and requires knowledge of growth rates, but Smith et al. (1992) revealed that a Armillaria bulbosa genet remained genetically stable over 1500 years. Interestingly, this fungus is also parasitic, illustrating that a fungus can survive longer than its host. In the case of ancient beech forests, fungal genets could be sampled to test if their age compared to recent forests, and therefore to determine if part of the diversity could be attributed to past conditions. A study of lichens and fungi associated with old oaks have revealed an in-between trend: their diversity was both shaped by past and recent conditions (Ranius et al., 2008a), but older oaks hosted the richer communities (Ranius et al., 2008a, b). Similarly, for plants, past fragmentation particularly affects the current distribution of only slow colonizing species (Vellend et al., 2006) causing a long-lasting extinction debt, which is possibly never paid (Dupouey et al., 2002). The same pattern may be true for fungi, demonstrating the need for further studies of fungal traits and especially their dispersal abilities in order to help understand differences of extinction debt observed between fungi (e.g., Berglund and Jonsson, 2005).
Beyond those distinct patterns for bacteria and fungi, our study has revealed new landscape effects on soil communities, and highlighted the interest of conserving large amount of ancient forests in the vicinity of recent forests to favor fungal diversity in soil. Interestingly, the results of a study of English forests by Spake et al. (2016) already demonstrate the benefit in conserving patches of ancient forests close to younger forests, when they detected a high similarity of fungal diversity and community composition between closely located pairs of forests, both dominated by oaks. All these results raise the question as to whether ancient forests could act as sources for fungal dispersal. The study of Bidartondo et al. (2001) in and near ancient forests of bristlecone pine revealed a low diversity of ectomycorrhizal inoculum, contrasting with the idea that old-growth forests would host more species, and at least “indicator” species, specifically associated with old trees. While the literature on fungal indicators is rich for wood-decaying fungi, results for ectomycorrhizal fungi are more contrasted. For example, Dahlberg et al. (1997) and Richard et al. (2005) have revealed a high diversity of ectomycorrhizal species in boreal and Mediterranean old-growth forests, respectively. A large-scale inventory in Czechia recently highlighted the correlation between ancient forests plants and fungal diversity, and suggested that ancient forest plants could be used to predict fungal richness, especially for red-listed species (Hofmeister et al., 2014), but rarely for ectomycorrhizal fungi. These results may depend on dominant tree species, as Boeraeve et al. (2018a) revealed that only late successional tree species host more diverse ectomycorrhizal communities in the oldest forests.
Our results, based on soil metabarcoding, suggest that nearly all fungi could be favored by greater coverage of ancient forest, and in particular highlight the strong effect on ectomycorrhizal fungi, dominant in our samples and more generally in beech-forest soils (Guinberteau, 2011; Coince et al., 2013). Using such techniques could therefore revolutionize the debate surrounding indicator species for ecological continuity (Rolstad et al., 2002) and allow for the comparison of diverse types of ancient forests, under different hosts. On the other hand, indicator species will still be useful for an above-ground diagnostic, since most sequences cannot be identified at the species level with metabarcoding techniques. For example, while Russulaceae and Cortinariaceae were the more abundant families, typical beech-associated fungi (mentioned by Guinberteau, 2011), such as Russula fageticola, R. faginea, R. rubroalba, R. mairei, Cortinarius montanus var. fageticola, and C. amoenolens were not detected with our short markers. Moreover, the cost of metabarcoding can remain high for a single inventory, and decreases only if several samples are collected (e.g., Zinger et al., 2016). Finally, if wood-decaying fungi are considered as good indicators, wood samples could be used to detect their presence with metabarcoding (e.g., Ovaskainen et al., 2010), and therefore make more comparisons with the existing literature on fungal diversity in ancient or old-growth forests than our study based on soil sampling.
Our study focused on soil microorganism diversity patterns, and revealed that both soil conditions and habitat coverage in a landscape shaped fungal diversity in the forests of the Cevennes National Park, and that bacterial communities were only shaped by soil conditions. While results on fungi are sometimes contradictory in the literature (Grilli et al., 2017), we detected a common pattern for all fungal guilds, showing how strong the effect of habitat coverage can be. Our results provide a synthetic view and show the importance of conserving any ancient forest patch, whatever its size, for soil biodiversity conservation. These results are of interest for park management but also for urban areas, where isolated patches of forest can have persisted over time and host unique fungi communities (Vogt-Schilb et al., 2018). Environmental sequencing could now be applied to produce comprehensive soil inventories, and study isolated systems, for example in urban areas or dry ecosystems, where patches of forests are particularly threatened and fungi could contribute to their functioning and connectivity, and even resilience when facing climate change (Gilliam, 2016).
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: DRYAD Repository (https://doi.org/10.5061/dryad.hmgqnk9c8).
Author Contributions
SMe, FK, MR, and SMa handled the field work. SMe and SMa did the extractions and molecular biology. FK handled the soil analysis. SMe and SMa did the bioinformatic analyses. SMe and AB did the statistical analyses. All authors contributed to discussion of the results and the writing of the final document.
Funding
This research was financially supported by FEDER, FNADT, and EI-PURPAN. We also benefitted from the support of “Investissements d’Avenir” grants managed by Agence Nationale de la Recherche (CEBA, ref. ANR-10-LABX-25-01; ANAEE-France: ANR-11-INBS-0001, TULIP: ANR-10-LABX-0041).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was conducted at the Cevennes National Park as part of the national action plan to preserve ancient forests and financially supported by FEDER, FNADT, and EI-PURPAN. We thank Mathieu Baconnet and Sophie Giraud for their advice, Patricia Jargeat for this help with DNA extraction and PCR, Francois Calatayud for his assistance with GIS, Michel Goulard and Magali San-Cristobal for their help with statistics. We finally thank Alain Roy and Julian Donald for reading our manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2020.00118/full#supplementary-material
Footnotes
- ^ ftp://ftp.ncbi.nlm.nih.gov/genbank
- ^ https://doi.org/10.5061/dryad.hmgqnk9c8
- ^ https://www6.hautsdefrance.inra.fr/las/
References
Abrego, N., and Salcedo, I. (2014). Response of wood-inhabiting fungal community to fragmentation in a beech forest landscape. Fungal Ecol. 8, 18–27.
Adams, R. I., Miletto, M., Taylor, J. W., and Bruns, T. D. (2013). Dispersal in microbes: fungi in indoor air are dominated by outdoor air and show dispersal limitation at short distances. ISME J. 7, 1262–1273. doi: 10.1038/ismej.2013.28
Alberdi, A., and Gilbert, M. T. P. (2019). hilldiv: an R package for the integral analysis of diversity based on Hill numbers. bioRxiv [Preprint]. doi: 10.1101/545665
Almasia, R., Carú, M., Handford, M., and Orlando, J. (2016). Environmental conditions shape soil bacterial community structure in a fragmented landscape. Soil Biol. Biochem. 103, 39–45.
Altieri, M. A. (1999). The ecological role of biodiversity in agroecosystems. Agric. Ecosyst. Environ. 74, 19–31.
Bani, A., Pioli, S., Ventura, M., Panzacchi, P., Borruso, L., Tognetti, R., et al. (2018). The role of microbial community in the decomposition of leaf litter and deadwood. Appl. Soil Ecol. 126, 75–84.
Bartoñ, K. (2019). Package ‘MuMIn’. Available online at: https://CRAN.R-project.org/package=MuMIn (accessed April 15, 2020).
Bates, D., Mächler, M., Bolker, B., and Walker, S. (2015). Fitting linear mixed-effects models using lme4. J. Stat. Softw. 67, 1–48. doi: 10.18637/jss.v067.i01
Bergès, L., Avon, C., Arnaudet, L., Archaux, F., Chauchard, S., and Dupouey, J.-L. (2016). Past landscape explains forest periphery-to-core gradient of understorey plant communities in a reforestation context. Divers. Distrib. 22, 3–16. doi: 10.1111/ddi.12384
Bergès, L., and Dupouey, J.-L. (2017). Écologie historique et ancienneté de l’état boisé: concepts, avancées et perspectives de la recherche. Revue Forestière Française 4–5, 297–318. doi: 10.4267/2042/67863
Berglund, H., and Jonsson, B. G. (2005). Verifying an extinction debt among lichens and fungi in northern Swedish boreal forests. Conserv. Biol. 19, 338–348. doi: 10.1111/j.1523-1739.2005.00550.x
Bidartondo, M. I., Baar, J., and Bruns, T. D. (2001). Low ectomycorrhizal inoculum potential and diversity from soils in and near ancient forests of bristlecone pine (Pinus longaeva). Can. J. Bot. 79, 293–299. doi: 10.1139/b01-006
Birkhofer, K., Schöning, I., Alt, F., Herold, N., Klarner, B., Maraun, M., et al. (2012). General relationships between abiotic soil properties and soil biota across spatial scales and different land-use types. PLoS One 7:e43292. doi: 10.1371/journal.pone.0043292
Boeraeve, M., Honnay, O., and Jacquemyn, H. (2018a). Effects of host species, environmental filtering and forest age on community assembly of ectomycorrhizal fungi in fragmented forests. Fungal Ecol. 36, 89–98.
Boeraeve, M., Honnay, O., Mullens, N., Vandekerkhove, K., De Keersmaeker, L., Thomaes, A., et al. (2018b). The impact of spatial isolation and local habitat conditions on colonization of recent forest stands by ectomycorrhizal fungi. For. Ecol. Manag. 429, 84–92.
Boyer, F., Mercier, C., Bonin, A., Le Bras, Y., Taberlet, P., and Coissac, E. (2016). obitools: a unix-inspired software package for DNA metabarcoding. Mol. Ecol. Resour. 16, 176–182. doi: 10.1111/1755-0998.12428
Brennan, J., Darren, J., Contreras, T. A., and Fahrig, L. (2002). “Focal patch landscape studies for wildlife management: optimizing sampling effort across scales,” in Integrating Landscape Ecology into Natural Resource Management, eds J. Liu and W. W. Taylor (New York, NY: Cambridge University Press), 68–91.
BRGM (2008). Carte Lithologiquesimplifiée ® [WGS raster].1: 1 000 000. Orléans: Bureau de Recherches Géologiques et Minières.
Burkey, T. V. (1997). Metapopulation extinction in fragmented landscapes: using bacteria and protozoa communities as model ecosystems. Am. Nat. 150, 568–591. doi: 10.1086/286082
Cateau, E., Larrieu, L., Vallauri, D., Savoie, J.-M., Touroult, J., and Brustel, H. (2015). Ancienneté et maturité: deux qualités complémentaires d’un écosystème forestier. C. R. Biol. 338, 58–73.
Chao, A., and Jost, L. (2012). Coverage-based rarefaction and extrapolation: standardizing samples by completeness rather than size. Ecology 93, 2533–2547. doi: 10.1890/11-1952.1
Chemidlin Prévost-Bouré, N., Dequiedt, S., Thioulouse, J., Lelièvre, M., Saby, N. P. A., Jolivet, C., et al. (2014). Similar processes but different environmental filters for soil bacterial and fungal community composition turnover on a broad spatial scale. PLoS One 9:e111667. doi: 10.1371/journal.pone.0111667
Christensen, M., Heilmann-Clausen, J., Walleyn, R., and Adamcik, S. (2005). “Wood-inhabiting fungi as indicators of nature value in European beech forests,” in Proceeding of the Monitoring and Indicators of Forest Biodiversity in Europe–From Ideas to Operationality, Florence, 229.
Coince, A., Caël, O., Bach, C., Lengellé, J., Cruaud, C., Gavory, F., et al. (2013). Below-ground fine-scale distribution and soil versus fine root detection of fungal and soil oomycete communities in a French beech forest. Fungal Ecol. 6, 223–235.
Culman, S. W., Young-Mathews, A., Hollander, A. D., Ferris, H., Sánchez-Moreno, S., O’Geen, A. T., et al. (2010). Biodiversity is associated with indicators of soil ecosystem functions over a landscape gradient of agricultural intensification. Landsc. Ecol. 25, 1333–1348.
Dahlberg, A., Jonsson, L., and Nylund, J.-E. (1997). Species diversity and distribution of biomass above and below ground among ectomycorrhizal fungi in an old-growth Norway spruce forest in south Sweden. Can. J. Bot. 75, 1323–1335. doi: 10.1139/b97-844
de Vries, F. T., Manning, P., Tallowin, J. R., Mortimer, S. R., Pilgrim, E. S., Harrison, K. A., et al. (2012). Abiotic drivers and plant traits explain landscape-scale patterns in soil microbial communities. Ecol. Lett. 15, 1230–1239. doi: 10.1111/j.1461-0248.2012.01844.x
Diedhiou, A. G., Dupouey, J.-L., Buee, M., Dambrine, E., Laüt, L., and Garbaye, J. (2009). Response of ectomycorrhizal communities to past Roman occupation in an oak forest. Soil Biol. Biochem. 41, 2206–2213.
Diedhiou, A. G., Dupouey, J.-L., Buée, M., Dambrine, E., Laüt, L., and Garbaye, J. (2010). The functional structure of ectomycorrhizal communities in an oak forest in central France witnesses ancient Gallo-Roman farming practices. Soil Biol. Biochem. 42, 860–862.
Douhan, G. W., Vincenot, L., Gryta, H., and Selosse, M.-A. (2011). Population genetics of ectomycorrhizal fungi: from current knowledge to emerging directions. Fungal Biol. 115, 569–597. doi: 10.1016/j.funbio.2011.03.005
Dray, S., Legendre, P., and Peres-Neto, P. R. (2006). Spatial modelling: a comprehensive framework for principal coordinate analysis of neighbour matrices (PCNM). Ecol. Model. 196, 483–493. doi: 10.1016/j.ecolmodel.2006.02.015
Dupouey, J. L., Dambrine, E., Laffite, J. D., and Moares, C. (2002). Irreversible impact of past land use on forest soils and biodiversity. Ecology 83, 2978–2984.
Dvořák, D., Vašutová, M., Hofmeister, J., Beran, M., Hošek, J., Běták, J., et al. (2017). Macrofungal diversity patterns in central European forests affirm the key importance of old-growth forests. Fungal Ecol. 27, 145–154. doi: 10.1016/j.funeco.2016.12.003
Edman, M., Kruys, N., and Jonsson, B. G. (2004). Local dispersal sources strongly affect colonization patterns of wood-decaying fungi on spruce logs. Ecol. Appl. 14, 893–901. doi: 10.1890/03-5103
Epp, L. S., Boessenkool, S., Bellemain, E. P., Haile, J., Esposito, A., Riaz, T., et al. (2012). New environmental metabarcodes for analysing soil DNA: potential for studying past and present ecosystems. Mol. Ecol. 21, 1821–1833. doi: 10.1111/j.1365-294X.2012.05537.x
Fahrig, L. (2013). Rethinking patch size and isolation effects: the habitat amount hypothesis. J. Biogeogr. 40, 1649–1663.
Ficetola, G., Coissac, E., Zundel, S., Riaz, T., Shehzad, W., Bessière, J., et al. (2010). An In silico approach for the evaluation of DNA barcodes. BMC Genomics 11:434. doi: 10.1186/1471-2164-11-434
Fierer, N., Breitbart, M., Nulton, J., Salamon, P., Lozupone, C., Jones, R., et al. (2007a). Metagenomic and small-subunit rRNA analyses reveal the genetic diversity of bacteria, archaea, fungi, and viruses in soil. Appl. Environ. Microbiol. 73, 7059–7066. doi: 10.1128/AEM.00358-07
Fierer, N., Morse, J. L., Berthrong, S. T., Bernhardt, E. S., and Jackson, R. B. (2007b). Environmental controls on the landscape-scale biogeography of stream bacterial communities. Ecology 88, 2162–2173. doi: 10.1890/06-1746.1
Fierer, N., and Jackson, R. B. (2006). The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. U.S.A. 103, 626–631. doi: 10.1073/pnas.0507535103
Fliegerova, K., Tapio, I., Bonin, A., Mrazek, J., Callegari, M. L., Bani, P., et al. (2014). Effect of DNA extraction and sample preservation method on rumen bacterial population. Anaerobe 29, 80–84. doi: 10.1016/j.anaerobe.2013.09.015
Gilbert, F., Gonzalez, A., and Evans-Freke, I. (1998). Corridors maintain species richness in the fragmented landscapes of a microecosystem. Proc. R. Soc. Lond. Ser. B Biol. Sci. 265, 577–582. doi: 10.1098/rspb.1998.0333
Gilliam, F. S. (2016). Forest ecosystems of temperate climatic regions: from ancient use to climate change. New Phytol. 212, 871–887. doi: 10.1111/nph.14255
Grilli, G., Longo, S., Huais, P. Y., Pereyra, M., Verga, E. G., Urcelay, C., et al. (2017). Fungal diversity at fragmented landscapes: synthesis and future perspectives. Curr. Opin. Microbiol. 37, 161–165. doi: 10.1016/j.mib.2017.09.003
Grilli, G., Urcelay, C., Galetto, L., Davison, J., Vasar, M., and Saks, Ü, et al. (2015). The composition of arbuscular mycorrhizal fungal communities in the roots of a ruderal forb is not related to the forest fragmentation process. Environ. Microbiol. 17, 2709–2720. doi: 10.1111/1462-2920.12623
Guinberteau, J. (2011). Contribution to the knowledge of the mycoflora of the beech grove relic of Ciron under a landais climate. Bull. Mycol. Botan Dauphiné Savoie 51, 23–38.
Halme, P., Holec, J., and Heilmann-Clausen, J. (2017). The history and future of fungi as biodiversity surrogates in forests. Fungal Ecol. 27, 193–201. doi: 10.1016/j.funeco.2016.10.005
Haddad, N. M., Gonzalez, A., Brudvig, L. A., Burt, M. A., Levey, D. J., and Damschen, E. I. (2017). Experimental evidence does not support the habitat amount Hypothesis. Ecography 40, 48–55. doi: 10.1111/ecog.02535
Harding, J. S., Benfield, E. F., Bolstad, P. V., Helfman, G. S., and Jones, E. B. D. (1998). Stream biodiversity: the ghost of land use past. Proc. Natl. Acad. Sci. U.S.A. 95, 14843–14847. doi: 10.1073/pnas.95.25.14843
Helander, M., Ahlholm, J., Sieber, T. N., Hinneri, S., and Saikkonen, K. (2007). Fragmented environment affects birch leaf endophytes. New Phytol. 175, 547–553. doi: 10.1111/j.1469-8137.2007.02110.x
Hermy, M., Honnay, O., Firbank, L., Grashof-Bokdam, C., and Lawesson, J. E. (1999). An ecological comparison between ancient and other forest plant species of Europe, and the implications for forest conservation. Biol. Conserv. 91, 9–22. doi: 10.1016/S0006-3207(99)00045-42
Hill, M. O. (1973). Diversity and evenness: a unifying notation and its consequences. Ecology 54, 427–432. doi: 10.2307/1934352
Hobara, S., Osono, T., Hirose, D., Noro, K., Hirota, M., and Benner, R. (2014). The roles of microorganisms in litter decomposition and soil formation. Biogeochemistry 118, 471–486.
Hofmeister, J., Hošek, J., Brabec, M., Dvořák, D., Beran, M., Deckerová, H., et al. (2014). Richness of ancient forest plant species indicates suitable habitats for macrofungi. Biodivers. Conserv. 23, 2015–2031.
Horner-Devine, M. C., Lage, M., Hughes, J. B., and Bohannan, B. J. (2004). A taxa–area relationship for bacteria. Nature 432:750.
Hsieh, T. C., Ma, K. H., and Chao, A. (2016). iNEXT: an R package for rarefaction and extrapolation of species diversity (H ill numbers). Methods Ecol. Evol. 7, 1451–1456. doi: 10.1111/2041-210X.12613
IGN (1996). BD ALTI ® [MNT]. 1: 25 000. Saint-Mandé: Institut National de l’information Géographique et forestière.
IGN (2011). SCAN Etat-Major® [TIF6.0]. 1: 40 000. Saint-Mandé: Institut National de l’information Géographique et forestière.
Jackson, H. B., and Fahrig, L. (2015). Are ecologists conducting research at the optimal scale: Is research conducted at optimal scales? Glob. Ecol. Biogeogr. 24, 52–63. doi: 10.1111/geb.12233
Jones, S. E., and Lennon, J. T. (2010). Dormancy contributes to the maintenance of microbial diversity. Proc. Natl. Acad. Sci. U.S.A. 107, 5881–5886. doi: 10.1073/pnas.0912765107
Kaprelyants, A., Gottschal, J. C., and Kell, D. B. (1993). Dormancy in non-sporulating bacteria. FEMS Microbiol. Lett. 104, 271–285. doi: 10.1016/0378-1097(93)90600-90607
Kuussaari, M., Bommarco, R., Heikkinen, R. K., Helm, A., Krauss, J., Lindborg, R., et al. (2009). Extinction debt: a challenge for biodiversity conservation. Trends Ecol. Evol. 24, 564–571. doi: 10.1016/j.tree.2009.04.011
Lauber, C. L., Hamady, M., Knight, R., and Fierer, N. (2009). Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 75, 5111–5120. doi: 10.1128/AEM.00335-339
Levis, C., Costa, F. R., Bongers, F., Peña-Claros, M., Clement, C. R., Junqueira, A. B., et al. (2017). Persistent effects of pre-Columbian plant domestication on Amazonian forest composition. Science 355, 925–931. doi: 10.1126/science.aal0157
Lindgren, J. P., and Cousins, S. A. (2017). Island biogeography theory outweighs habitat amount hypothesis in predicting plant species richness in small grassland remnants. Landsc. Ecol. 32, 1895–1906.
Lucas, R., Groeneveld, J., Harms, H., Johst, K., Frank, K., and Kleinsteuber, S. (2017). A critical evaluation of ecological indices for the comparative analysis of microbial communities based on molecular datasets. FEMS Microbiol. Ecol. 93:fiw209. doi: 10.1093/femsec/fiw209
Martin, C. A. (2018). An early synthesis of the habitat amount hypothesis. Landsc. Ecol. 33, 1831–1835. doi: 10.1007/s10980-018-0716-y
Melo, G. L., Sponchiado, J., Cáceres, N. C., and Fahrig, L. (2017). Testing the habitat amount hypothesis for South American small mammals. Biol. Conserv. 209, 304–314.
Nagati, M., Roy, M., Manzi, S., Richard, F., Desrochers, A., Gardes, M., et al. (2018). Impact of local forest composition on soil fungal communities in a mixed boreal forest. Plant Soil 432, 345–357.
Nguyen, N. H., Song, Z., Bates, S. T., Branco, S., Tedersoo, L., Menke, J., et al. (2016). FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 20, 241–248.
Nilsson, R. H., Kristiansson, E., Ryberg, M., Hallenberg, N., and Larsson, K.-H. (2008). Intraspecific ITS variability in the kingdom Fungi as expressed in the international sequence databases and its implications for molecular species identification. Evol. Bioinformatics 4, 193–201. doi: 10.4137/ebo.s653
Nordén, B., Dahlberg, A., Brandrud, T. E., Fritz, Ö, Ejrnaes, R., and Ovaskainen, O. (2014). Effects of ecological continuity on species richness and composition in forests and woodlands: a review. Écoscience 21, 34–45. doi: 10.2980/21-1-3667
Nordén, J., Åström, J., Josefsson, T., Blumentrath, S., Ovaskainen, O., Sverdrup-Thygeson, A., et al. (2018). At which spatial and temporal scales can fungi indicate habitat connectivity? Ecol. Indic. 91, 138–148. doi: 10.1186/s13071-017-2482-0
Nordén, J., Penttilä, R., Siitonen, J., Tomppo, E., and Ovaskainen, O. (2013). Specialist species of wood-inhabiting fungi struggle while generalists thrive in fragmented boreal forests. J. Ecol. 101, 701–712.
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O’hara, R. B., et al. (2019). vegan: Community Ecology Package. R Package Version 2.5–6. Available online at: http://CRAN.R-project.org/package=vegan (accessed September 1, 2019).
Ovaskainen, O., Nokso-Koivisto, J., Hottola, J., Rajala, T., Pennanen, T., Ali-Kovero, H., et al. (2010). Identifying wood-inhabiting fungi with 454 sequencing–what is the probability that BLAST gives the correct species? Fungal Ecol. 3, 274–283.
Parmasto, E. (2001). “Fungi as indicators of primeval and old-growth forests deserving protection,” in Fungal Conservation Issues and Solutions, eds D. Moore, M. M. Nauta, S. E. Evans, and M. Rotheroe (Cambridge: Cambridge University Press), 81–88.
Peay, K. G., Schubert, M. G., Nguyen, N. H., and Bruns, T. D. (2012). Measuring ectomycorrhizal fungal dispersal: macroecological patterns driven by microscopic propagules. Mol. Ecol. 21, 4122–4136. doi: 10.1111/j.1365-294X.2012.05666.x
Pozzi, A. C., Bautista-Guerrero, H. H., Nouioui, I., Cotin-Galvan, L., Pepin, R., Fournier, P., et al. (2015). In-planta sporulation phenotype: a major life history trait to understand the evolution of A lnus-infective F rankia strains. Environ. Microbiol. 17, 3125–3138. doi: 10.1111/1462-2920.12644
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2012). The SILVA ribosomal RNA genedatabaseproject: improved data processing and web-basedtools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
R Core Team (2019). A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Ranius, T., Eliasson, P., and Johansson, P. (2008a). Large-scale occurrence patterns of red-listed lichens and fungi on old oaks are influenced both by current and historical habitat density. Biodivers. Conserv. 17, 2371–2381.
Ranius, T., Johansson, P., Berg, N., and Niklasson, M. (2008b). The influence of tree age and microhabitat quality on the occurrence of crustose lichens associated with old oaks. J. Veg. Sci. 19, 653–662. doi: 10.3170/2008-8-18433
Richard, F., Millot, S., Gardes, M., and Selosse, M.-A. (2005). Diversity and specificity of ectomycorrhizal fungi retrieved from an old-growth Mediterranean forest dominated by Quercus ilex. New Phytol. 166, 1011–1023. doi: 10.1111/j.1469-8137.2005.01382.x
Rolstad, J., Gjerde, I., Gundersen, V. S., and Saetersdal, M. (2002). Use of indicator species to assess forest continuity: a critique. Conserv. Biol. 16, 253–257. doi: 10.1046/j.1523-1739.2002.00552.x
Schrott, G. R., With, K. A., and King, A. W. (2005). Demographic limitations of the ability of habitat restoration to rescue declining populations: demographic limits of habitat restoration. Conserv. Biol. 19, 1181–1193. doi: 10.1111/j.1523-1739.2005.00205.x
Seibold, S., Bässler, C., Brandl, R., Fahrig, L., Förster, B., Heurich, M., et al. (2017). An experimental test of the habitat-amount hypothesis for saproxylic beetles in a forested region. Ecology 98, 1613–1622. doi: 10.1002/ecy.1819
Smith, D. J., Timonen, H. J., Jaffe, D. A., Griffin, D. W., Birmele, M. N., Perry, K. D., et al. (2013). Intercontinental dispersal of bacteria and archaea by transpacific winds. Appl. Environ. Microbiol. 79, 1134–1139. doi: 10.1128/AEM.03029-12
Smith, M. L., Bruhn, J. N., and Anderson, J. B. (1992). The fungus Armillaria bulbosa is among the largest and oldest living organisms. Nature 356, 428–431. doi: 10.1038/356428a0
Spake, R., van der Linde, S., Newton, A. C., Suz, L. M., Bidartondo, M. I., and Doncaster, C. P. (2016). Similar biodiversity of ectomycorrhizal fungi in set-aside plantations and ancient old-growth broadleaved forests. Biol. Conserv. 194, 71–79. doi: 10.1016/j.biocon.2015.12.003
Taberlet, P., Bonin, A., Zinger, L., and Coissac, E. (2018). Environmental DNA: For Biodiversity Research and Monitoring. Oxford: Oxford University Press.
Taberlet, P., Coissac, E., Pompanon, F., Brochmann, C., and Willerslev, E. (2012). Towards next-generation biodiversity assessment using DNA metabarcoding. Mol. Ecol. 21, 2045–2050. doi: 10.1111/j.1365-294X.2012.05470.x
Vacher, C., Cordier, T., and Vallance, J. (2016). Phyllosphere fungal communities differentiate more thoroughly than bacterial communities along an elevation gradient. Microb. Ecol. 72, 1–3. doi: 10.1007/s00248-016-0742-748
Vannette, R. L., Leopold, D. R., and Fukami, T. (2016). Forest area and connectivity influence root-associated fungal communities in a fragmented landscape. Ecology 97, 2374–2383. doi: 10.1002/ecy.1472
Vellend, M., Verheyen, K., Jacquemyn, H., Kolb, A., Van Calster, H., Peterken, G., et al. (2006). Extinction debt of forest plants persists for more than a century following habitat fragmentation. Ecology 87, 542–548. doi: 10.1890/05-1182
Vincenot, L., and Selosse, M.-A. (2017). “Population biology and ecology of ectomycorrhizal fungi,” in Biogeography of Mycorrhizal Symbiosis, ed. L. Tedersoo (Cham: Springer International Publishing AG), 39–59.
Vogt-Schilb, H., Moreau, P.-A., Malaval, J.-C., Schatz, B., and Richard, F. (2018). Effects of long-term landscape dynamics and city growth on biodiversity in the Mediterranean: the case study of Montpellier, France. Urban Ecosyst. 21, 921–932. doi: 10.1007/s11252-018-0780-788
Keywords: landscape ecology, habitat amount hypothesis, fungi, bacteria, metabarcoding
Citation: Mennicken S, Kondratow F, Buralli F, Manzi S, Andrieu E, Roy M and Brin A (2020) Effects of Past and Present-Day Landscape Structure on Forest Soil Microorganisms. Front. Ecol. Evol. 8:118. doi: 10.3389/fevo.2020.00118
Received: 11 December 2019; Accepted: 14 April 2020;
Published: 19 May 2020.
Edited by:
Cendrine Mony, University of Rennes 1, FranceReviewed by:
Juan Ernesto Guevara Andino, Field Museum of Natural History, United StatesMargaux Boeraeve, KU Leuven, Belgium
Copyright © 2020 Mennicken, Kondratow, Buralli, Manzi, Andrieu, Roy and Brin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mélanie Roy, melanie.roy@univ-tlse3.fr